Zulassung & Anmeldung von Medizinprodukten
Mit Sicherheit zum Ziel – Ihr Weg zur CE-Kennzeichnung
Die Zulassung von Medizinprodukten in Europa ist kein einfacher Weg – aber ein notwendiger. Zwischen MDR, IVDR, CE-Kennzeichnung und EUDAMED verlieren viele Hersteller den Überblick. Wir helfen Ihnen, diesen Weg sicher und effizient zu gehen.
Typische Hürden bei der Medizinprodukte-Zulassung
Unterstützung von A-Z: Bis Ihr Medizinprodukt am Markt ist

- Viele Vorgaben aus MDR (EU 2017/745) und IVDR bei wenig Zeit
- Unsicherheit bei Klassifizierung/Risikoklasse und dem passenden Konformitätsbewertungsverfahren
- Fragen zu Technischer Dokumentation, klinischen Daten, ISO 13485 und zur Einbindung einer Benannten Stelle
Wir sorgen für Planbarkeit, Tempo und Sicherheit.
Ihr Partner für das Inverkehrbringen von Medizinprodukten
Von der Idee bis zur Zulassung: Begleitung für Ihr Medizinprodukt
Ob Start-up oder etablierter Hersteller – wir stehen Ihnen zur Seite, wenn es darum geht, Ihr Medizinprodukt sicher, effizient und gesetzeskonform auf den europäischen Markt zu bringen.
Mit maßgeschneiderten Lösungen für jede Projektphase begleiten wir Sie ganzheitlich – vom ersten Konzept bis zur erfolgreichen Zulassung.
Unsere Leistungen für Ihre MDR/IVDR-konforme Medizinprodukte-Zulassung
Wir begleiten Sie durch den gesamten Zulassungsprozess
Wir denken nicht nur einzelne Schritte – wir denken den gesamten Prozess mit. Ob Klassifizierung, technische Dokumentation, QMS oder Kommunikation mit Behörden: Wir sind an Ihrer Seite – bis Ihr Produkt sicher auf dem Markt ist.
Zulassungsvoraussetzungen für Hersteller von Medizinprodukten
Das müssen Sie mitbringen
Sie möchten Ihr Medizinprodukt oder Ihre IVD schnell, sicher und rechtskonform auf den europäischen Markt bringen?
Wir begleiten Sie dabei – mit einem klaren Blick auf das Wesentliche und einem strukturierten Fahrplan zur CE-Kennzeichnung.
Damit Ihr Produkt erfolgreich die CE-Kennzeichnung erhält und in der EU in Verkehr gebracht werden darf, müssen bestimmte Voraussetzungen erfüllt sein.
Hersteller müssen die Anforderungen der MDR bzw. IVDR vollständig umsetzen – wir zeigen Ihnen, worauf es wirklich ankommt.
Ihre To-do-Liste für die Zulassung:
- Ein Qualitätsmanagementsystem nach DIN EN ISO 13485 oder gemäß MDR / IVDR
- Eine vollständige technische Dokumentation nach Anhang II und III – strukturiert, prüfbereit, nachvollziehbar
- Ein systematisches Risikomanagement nach ISO 14971 - über den gesamten Produktlebenszyklus hinweg
- Eine klinische Bewertung oder ggf. klinische Prüfung, um Sicherheit und Leistung Ihres Produkts nachzuweisen
- Eine verantwortliche Person gemäß Artikel 15 MDR/IVDR - mit nachgewiesener Fachkompetenz
- Alle erforderlichen Nachweise und Aufzeichnungen, wie sie in der jeweiligen Verordnung gefordert sind
Gut zu wissen: Je besser diese Grundlagen vorbereitet sind, desto reibungsloser verläuft das Konformitätsbewertungsverfahren – und desto schneller kommt Ihr Produkt auf den Markt.
Risikoklassen von Medizinprodukten
Klassifizierung leicht erklärt
l
Risikoklasse l
Geringes Risiko für Patienten und Anwendern
Beispiel: Verbandmittel, Lesebrillen
Is / Im / Ir
Risikoklasse Is / Im / Ir
Geringes Risiko mit besonderen Anforderungen (z. B. steril, messend, wiederverwendbar)
Beispiel: Sterile Tupfer, Thermometer, Skalpelle
IIa
Risikoklasse IIa
Mittleres Risiko – meist nicht-invasiv oder kurzzeitig invasiv
Beispiel: Zahnfüllmaterial, Hörgeräte
IIb
Risikoklasse IIb
Hohes Risiko – z. B. langzeitig invasiv, implantierbar oder lebensunterstützend
Beispiel: Infusionspumpen, Beatmungsgeräte
III
Risikoklasse III
Sehr hohes Risiko – z. B. lebenswichtige oder dauerhaft implantierte Produkte
Beispiel: Herzklappen, Implantate
Medizinprodukte werden in vier Risikoklassen eingeteilt: I, IIa, IIb und III. Je höher die Klasse, desto strenger die Anforderungen. Diese Einstufung entscheidet darüber, ob Sie Ihr Produkt selbst deklarieren dürfen oder eine Benannte Stelle einbezogen werden muss.
Verfahrensaufwand je Risikoklasse
Klasse I
→ Selbstdeklaration durch den Hersteller
Klasse Is / Im / Ir
→ Prüfung des Sonderaspekts (z. B. Sterilität, Messfunktion) durch eine Benannte Stelle
Klasse IIa, IIb, III
→ Prüfung der technischen Dokumentation
→ Audit des Qualitätsmanagementsystems (ISO 13485)
→ Bewertung der klinischen Daten
→ Ausstellung eines Zertifikats durch die Benannte Stelle
Unser Tipp: Eine gründliche Klassifizierung spart Zeit, Kosten und vermeidet Rückfragen durch Behörden oder Benannte Stellen. In unseren FAQs finden Sie konkrete Produktbeispiele je Klasse.
Bereit für den Markt
Was vor der Markteinführung zählt
Bevor Ihr Medizinprodukt oder Ihre IVD in der EU in Verkehr gebracht werden darf, müssen alle regulatorischen Voraussetzungen erfüllt sein.
Wir helfen Ihnen, diese letzten Schritte sicher und vollständig umzusetzen – damit Ihr Produkt rechtskonform und erfolgreich auf den Markt kommt.
Was Sie benötigen:
- Erfolgreich abgeschlossenes Konformitätsbewertungsverfahren
- CE-Kennzeichnung am Produkt (inkl. Kennnummer der Benannten Stelle, falls erforderlich)
- Registrierung in EUDAMED für Hersteller, Produkte und verantwortliche Personen
- Funktionierendes Post-Market Surveillance (PMS)-System
- Vigilanzsystem zur Meldung schwerwiegender Vorkommnisse (z. B. an das BfArM)
Behördliche Schritte zur Zulassung in der EU
Die 9 Schritte zur Marktzulassung
Die Anforderungen sind in der MDR (EU 2017/745) bzw. IVDR geregelt. Je nach Risikoklasse sind unterschiedliche Schritte notwendig – insbesondere bei Produkten der Klassen IIa, IIb und III.
Wir begleiten Sie durch jeden Schritt – rechtssicher, effizient und mit Weitblick.
1
Klassifizierung des Produkts
- Einstufung gemäß MDR oder IVDR
- Bei Unsicherheiten: Vorabklärung mit dem BfArM möglich
2
Auswahl der Benannten Stelle (falls erforderlich)
- Ab Klasse Is/Im/Ir: Einbindung einer Konformitätsbewertungsstelle
3
Einreichung der technischen Unterlagen
- Zweckbestimmung, Risikomanagementakte, Gebrauchsanweisung, klinische Bewertung, Design- und Herstellungsdaten
Audit des Qualitätsmanagementsystems
- Nachweis eines funktionierenden QMS nach ISO 13485
5
5
Bewertung der klinischen Daten
- Prüfung der klinischen Bewertung oder klinischen Prüfung
- Genehmigung durch Ethikkommission und ggf. BfArM
6
4
Ausstellung des Zertifikats
- Durch die Benannte Stelle – Voraussetzung für die CE-Kennzeichnung
7
EU-Konformitätserklärung & CE-Kennzeichnung
- Offizielle Erklärung der Konformität mit MDR/IVDR
- CE-Zeichen mit vierstelliger Kennnummer der Benannten Stelle (bei höheren Klassen)
8
9
Registrierung in EUDAMED
- Eintrag von Hersteller, Produkt und verantwortlicher Person
- Dient der Marktüberwachung durch die Behörden
Post-Market Surveillance & Vigilanz
- Einrichtung eines Systems zur Marktbeobachtung
- Meldung schwerwiegender Vorkommnisse an Behörden
Unser Tipp: Je früher Sie mit der Planung beginnen, desto reibungsloser verläuft Ihre Zulassung.
Nächster Schritt - sprechen wir über Ihr QMS
Wir wissen:
Jedes Unternehmen ist einzigartig. Deshalb beginnen wir mit einem persönlichen Gespräch, um Ihre Anforderungen genau zu verstehen.
Konformitätsbewertung
Ihr Fahrplan zur Zulassung
Die Konformitätsbewertung ist das Herzstück Ihrer Zulassung. Je nach Risikoklasse gelten unterschiedliche Verfahren – von der einfachen Selbstdeklaration bis zur umfassenden Prüfung durch eine Benannte Stelle. Wir zeigen Ihnen, welches Verfahren für Ihr Produkt passt und begleiten Sie Schritt für Schritt.
CE-Kennzeichnung
Der letzte Schritt vor dem Marktzugang
Mit der CE-Kennzeichnung bestätigen Sie, dass Ihr Produkt alle EU-Anforderungen erfüllt. Dazu gehört die Konformitätserklärung, die technische Dokumentation und – je nach Klasse – die Einbindung einer Benannten Stelle. Wir sorgen dafür, dass Sie sicher und effizient zum Ziel kommen.
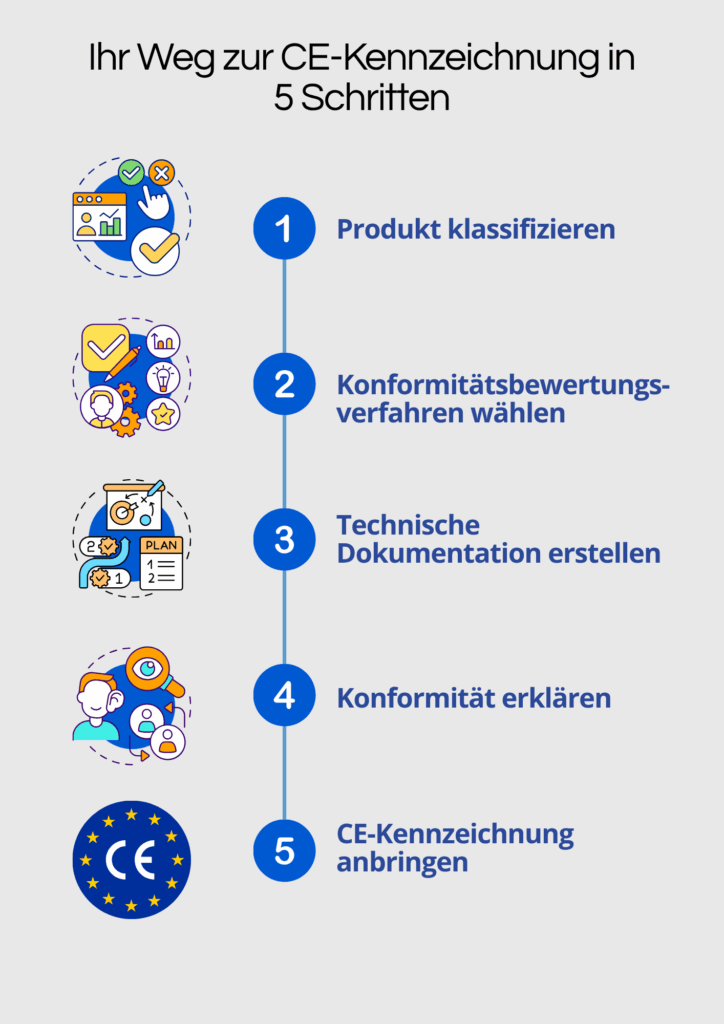
Was Sie als Hersteller wissen müssen
- Die CE-Kennzeichnung wird nicht beantragt, sondern vom Hersteller selbst angebracht – auf Basis einer erfolgreichen Konformitätsbewertung.
- Bei Produkten der Klassen Is, Im, Ir, IIa, IIb oder III muss zusätzlich die vierstellige Kennnummer der Benannten Stelle neben dem CE-Zeichen stehen.
- Die Kennzeichnung muss gut sichtbar, lesbar und dauerhaft auf dem Produkt oder der Verpackung angebracht sein.
EU-Konformitätserklärung
Bevor Sie das CE-Zeichen anbringen dürfen, müssen Sie als Hersteller eine EU-Konformitätserklärung ausstellen.
Darin bestätigen Sie, dass Ihr Produkt alle grundlegenden Sicherheits- und Leistungsanforderungen erfüllt – gemäß MDR oder IVDR.
Unser Hinweis: Die CE-Kennzeichnung ist mehr als ein Symbol, sie ist Ihr offizieller Nachweis für Sicherheit, Qualität und Rechtskonformität.
Europäische Datenbank für Medizinprodukte
EUDAMED
Seit dem 26. Mai 2021 gilt: Alle Medizinprodukte, die in der EU in Verkehr gebracht werden, müssen in EUDAMED registriert sein. Diese zentrale Datenbank sammelt alle relevanten Informationen zu Medizinprodukten – transparent, zugänglich und sicher.
Was ist Ziel von EUDAMED?
1. Mehr Sicherheit für Patienten und Anwender:
EUDAMED stärkt die Marktüberwachung und schafft Klarheit. Behörden und zuständige Stellen erhalten einen umfassenden Überblick über alle zugelassenen Produkte – für mehr Vertrauen und Sicherheit.
2. Freier Warenverkehr in der EU:
Durch die zentrale Bereitstellung von Produktinformationen erleichtert EUDAMED den Austausch zwischen Herstellern, Behörden und anderen Beteiligten – und fördert so den reibungslosen Handel innerhalb Europas.
EUDAMED ist damit ein Schlüsselinstrument für Transparenz, Sicherheit und Zusammenarbeit im europäischen Medizinproduktemarkt.
Wichtiger Hinweis
Der Weg zur Marktzulassung kann je nach Produkt und gewähltem Konformitätsbewertungsverfahren unterschiedlich sein. Hersteller sollten frühzeitig fachliche Unterstützung einholen – etwa durch spezialisierte Beratungsfirmen oder Rechtsanwälte im Bereich Regulatory Affairs –, um den Prozess sicher und effizient zu gestalten.
Häufig gestellte Fragen zur Zulassung von Medizinprodukten
FAQ
Die Zweckbestimmung entscheidet: Dient Ihr Produkt der Diagnose, Prävention, Überwachung oder Behandlung von Krankheiten? Dann fällt es unter die MDR oder IVDR. Wir helfen Ihnen bei der Einstufung.
Die Risikoklasse (I, IIa, IIb, III) bestimmt, wie aufwendig das Zulassungsverfahren ist. Je höher das Risiko, desto strenger die Anforderungen. Eine korrekte Klassifizierung spart Zeit und Kosten.
| Risikoklasse | Beispielprodukte |
|---|---|
| Klasse I | Verbandmaterial, Lesebrillen |
| Klasse Is | Sterile Einmalspritzen |
| Klasse IIa | Zahnfüllmaterial, Hörgeräte |
| Klasse IIb | Infusionspumpen, Beatmungsgeräte |
| Klasse III | Herzklappen, implantierbare Defibrillatoren |
Die CE-Kennzeichnung ist das sichtbare Zeichen für die Einhaltung der EU-Vorgaben. Sie erhalten sie nach erfolgreicher Konformitätserklärung und – je nach Klasse – Prüfung durch eine Benannte Stelle.
Ja – die Registrierung in der europäischen Datenbank EUDAMED ist verpflichtend. Sie sorgt für Transparenz und Rückverfolgbarkeit. Wir übernehmen die Anmeldung für Sie oder zeigen Ihnen, wie es geht.
Haben Sie Fragen?
Sie interessieren sich für eine Beratung oder eine Schulung und möchten sich dafür mit uns in Verbindung setzen?
Vereinbaren Sie gerne einen Termin für ein Gespräch : Kostenlos und unverbindlich.
Beratung: +49 34293 4797-20
Schulungen: +49 34293 4797-27