Technische Dokumentation Medizinprodukte
gemäß MDR / IVDR - konform, strukturiert und auditfest
Sie benötigen eine technische Dokumentation gemäß MDR oder IVDR, die alle Anforderungen der Medical Device Regulation erfüllt – klar gegliedert, vollständig und auditbereit? Ob für die Zulassung von Medizinprodukten, die Überwachung nach dem Inverkehrbringen oder die Vorbereitung auf die Benannte Stelle.
Wir unterstützen Sie bei der Erstellung der technischen Dokumentation – modular, effizient und nachvollziehbar.
Technische Dokumentation für Medizinprodukte
Starten Sie jetzt mit klarer Struktur für Ihre Nachweise
Normen, Nachweise, Deadlines – und dazwischen der Alltag. MDR, IVDR, Anhang I (GSPR), Anhang II/III, MDCG-Guidances, Benannte Stelle: Die Anforderungen sind komplex und verbindlich. Häufig sind Informationen verteilt – Entwicklung, Risikoanalyse, Usability, klinische Bewertung, PMS.
Wir gliedern Ihre technische Dokumentation für Medizinprodukte klar, bündeln Inhalte und schaffen nachvollziehbare Nachweise – gemäß MDR / IVDR und ISO 13485.
Unsere Leistungen rund um die Technische Dokumentation
gemäß MDR/IVDR
Wir unterstützen Sie gezielt bei der Erstellung, Überarbeitung und Strukturierung Ihrer Technischen Dokumentation für Medizinprodukte – mit Fokus auf Regelkonformität, Nachvollziehbarkeit und Auditfestigkeit.
Ihre Vorteile
spürbar im Alltag und bei Audits
-
Sicher durch den Regulierungsdschungel
Sie behalten den Überblick über MDR/IVDR-Anforderungen - ohne sich durch komplexe Anhänge und Leitlinien kämpfen zu müssen. -
Mehr Klarheit, weniger Unsicherheit
Unsere systematische Herangehensweise schafft Ordnung in der Dokumentation und reduziert Unsicherheiten bei Audits und Behördenanfragen. -
Zeitgewinn für Ihre Kernaufgaben
Sie konzentrieren sich auf Produktentwicklung und Marktstrategie - wir unterstützen Sie bei der Dokumentationsstruktur und Bewertung. -
Stärkung Ihrer internen Kompetenz
Durch gemeinsame Erarbeitung und Beratung bleibt das Wissen im Unternehmen - ideal für zukünftige Updates und interne Schulungen. -
Flexibilität nach Ihrem Bedarf
Ob punktuelle Unterstützung oder Begleitung bis zur CE-Kennzeichnung: Sie entscheiden, wie viel externe Hilfe Sie benötigen.
Wer profitiert von uns
Wir verstehen Ihre Branche - und Ihre Anforderungen
- Hersteller von Medizinprodukten (vom Start-up bis zum Konzern)
- Eigenmarken
- Inverkehrbringer
- Hersteller von Software als Medizinprodukt
- IVDR-Produzenten (Reagenz, Kits, Geräte)
Lassen Sie uns gemeinsam Ihre Risiken beherrschen
Häufig gestellte Fragen zur Technischen Dokumentation für Medizinprodukte
FAQ
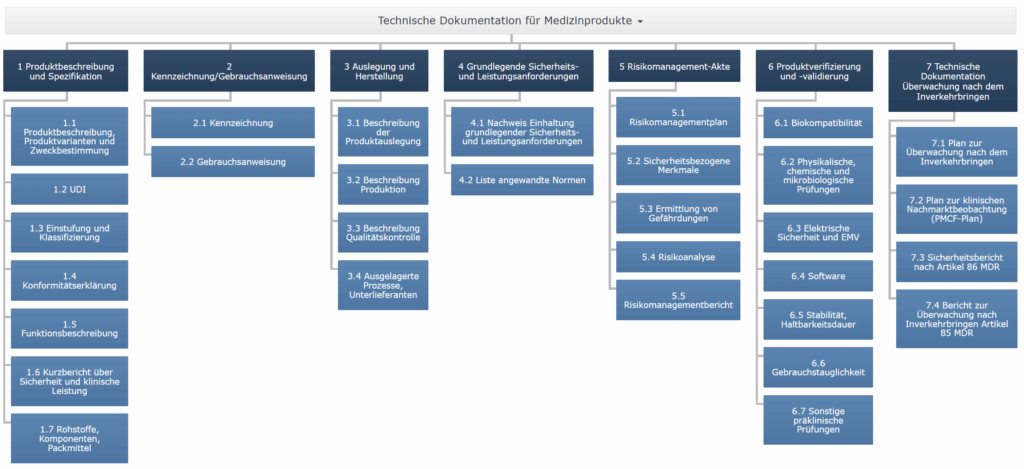
Die Technische Dokumentation (auch „Technical File“) umfasst alle Unterlagen, die ein Hersteller benötigt, um die Konformität seines Medizinprodukts mit der Medical Device Regulation (MDR) oder der In-vitro Diagnostic Regulation (IVDR) nachzuweisen. Sie ist Voraussetzung für die CE-Kennzeichnung und die Zulassung des Produkts in der EU.
Die MDR nennt in Anhang II und III die wesentlichen Anforderungen an die Technische Dokumentation. Dazu gehören u. a.:
- Produktbeschreibung und Spezifikationen
- Klassifizierung und Zweckbestimmung
- Design- und Herstellungsinformationen
- Sicherheits- und Leistungsanforderungen (GSPR)
- Risikoanalyse gemäß ISO 14971
- Klinische Bewertung bzw. Leistungsbewertung
- Gebrauchsanweisung und Etikettierung
- PMS-/PMCF-Dokumente (Post-Market Surveillance)
- Konformitätserklärung
GSPR steht für „General Safety and Performance Requirements“ – also die grundlegenden Sicherheits- und Leistungsanforderungen. Diese sind in Anhang I der Medizinprodukteverordnung definiert und müssen durch die Technische Dokumentation vollständig und nachvollziehbar erfüllt werden.
STED steht für Summary Technical Documentation und ist ein Strukturmodell, das vom IMDRF (International Medical Device Regulators Forum) empfohlen wird. Es hilft, die Technische Dokumentation klar, organisiert und durchsuchbar zu gestalten – besonders hilfreich bei internationalen Zulassungen.
Die MDR fordert, dass die Technische Dokumentation laufend aktualisiert wird – insbesondere bei Änderungen am Produkt, neuen Erkenntnissen aus der Marktüberwachung (PMS) oder bei regulatorischen Anpassungen. Eine regelmäßige GAP-Analyse hilft, Lücken frühzeitig zu erkennen.
Laut Artikel 15 MDR muss jedes Unternehmen eine „Qualified Person“ benennen, die für die Einhaltung der regulatorischen Anforderungen verantwortlich ist – einschließlich der Erstellung und Pflege der Technischen Dokumentation.
Unvollständige oder fehlerhafte Dokumentation kann zur Ablehnung durch die Benannte Stelle, zu Auditabweichungen oder sogar zum Marktrückruf führen. Eine strukturierte Erstellung und regelmäßige Prüfung sind daher essenziell.
Die Dokumentation muss in einer Sprache vorliegen, die von der Benannten Stelle und der zuständigen Behörde verstanden wird – meist Deutsch oder Englisch. Gebrauchsanweisungen (IFU) müssen zusätzlich marktgerecht übersetzt sein.
Haben Sie Fragen?
Sie interessieren sich für eine Beratung oder eine Schulung und möchten sich dafür mit uns in Verbindung setzen?
Vereinbaren Sie gerne einen Termin für ein Gespräch : Kostenlos und unverbindlich.
Beratung: +49 34293 4797-20
Schulungen: +49 34293 4797-27